VALERIE JEANNERET LOPEZ
Estudiante Medicina IX semestre
Universidad de los Andes
DANIELA ABRIL MELGAREJO
Estudiante Medicina VIII semestre
Universidad de los Andes
DEYANIRA GONZALEZ DEVIA
MD Especialista en Medicina Interna y Endocrinología
Fundación Santa Fe de Bogotá - Universidad de los Andes
La cirrosis se define como un estado avanzado de fibrosis hepática que se caracteriza por pérdida de los hepatocitos, destrucción de la arquitectura hepática, proliferación de miofibroblastos hepáticos y depósito de matriz extracelular incrementada. Esto conlleva a detoxificación insuficiente, carcinoma hepatocelular, hipertensión portal, falla renal y pulmonar, asociándose con incremento de la mortalidad.
Un evento clave en la fibrosis hepática es la activación y proliferación de las células hepáticas estrelladas. Las células hepáticas quiescentes almacenan vitamina A y residen en el espacio subendotelial de Disse. La lesión hepática crónica conlleva a la activación de estas células, convirtiéndose en contráctiles, produciendo componentes de matriz extracelular y secretando citoquinas y quemoquinas proinflamatorias tales como el factor de crecimiento transformante beta (1).
La activación de la célula hepática estrellada depende de las células de Kupffer, de las células endoteliales, hepatocitos y plaquetas. El depósito de matriz extracelular es constantemente opuesto a la degradación de estas proteínas. En la fibrosis hepática progresiva el balance se desvía hacia el depósito de matriz extracelular excesiva. Las metaloproteinasas de la matriz y sus reguladores (inhibidores tisulares de las metaloproteinasas, TIMPs) controlan el depósito y degradación de matriz. En la fibrogénesis hepática, TIMP-1 es producido por las células hepáticas estrelladas (2).
Múltiples situaciones pueden desencadenar una cirrosis, entre ellas infecciones virales, enfermedades de depósito, enfermedades autoinmunes, otras enfermedades metabólicas y medicamentos.
En cuanto a la fisiopatología de la enfermedad ósea en el paciente con cirrosis, podemos decir que se trata de un evento multifactorial, y que está asociado a factores iatrogénicos, como el uso de medicamentos, así como también a la osteodistrofia hepática preexistente. Múltiples estudios de corte transversal han demostrado una reducción en la masa ósea y alta prevalencia de osteoporosis en pacientes en valoración para trasplante hepático.
EPIDEMIOLOGÍA
La literatura reporta una prevalencia de osteoporosis en pacientes con cirrosis que varía entre el 12 y el 55% (3). Existen diversas razones para la amplia diferencia en la prevalencia encontrada en los estudios, atribuida a las desigualdades en la edad de la población incluida, el estado gonadal, la duración de la enfermedad, la severidad de la disfunción hepática y el desconocimiento de exposición a tratamiento para la enfermedad ósea.
En cuanto a la prevalencia de fracturas en la enfermedad hepática crónica, se ha encontrado igualmente información variable. La prevalencia oscila entre el 7 y el 35% para fracturas de cualquier característica y entre el 3 y el 44% para fracturas vertebrales (4,5). Las fracturas de cadera son muy raras debido a que la mayoría de pacientes con enfermedad hepática crónica no sobreviven hasta ser adultos mayores (6). La cirrosis aumenta en un factor de 2 el riesgo de presentar una fractura, así mismo, la prevalencia de fracturas incrementa con la edad. Se ha encontrado una tasa de fracturas en pacientes con cirrosis postmenopáusicas del 67%, en hombres 15% y en mujeres jóvenes del 6 -20% (4). La presencia de fracturas vertebrales se relaciona con una Densidad Mineral Osea (DMO) en columna lumbar baja, con la severidad de la disfunción hepática y con el hipogonadismo. Las fracturas periféricas se correlacionan con la presencia de cirrosis, el hipogonadismo y el consumo de alcohol (4).
FRAGILIDAD ÓSEA EN EL PACIENTE CON CIRROSIS
La cirrosis establecida se asocia a baja densidad ósea cuando se compara con enfermedad hepática no cirrótica. La duración de la enfermedad hepática no se correlaciona con la masa ósea; sin embargo, los estados cirróticos avanzados clínicos e histológicos presentan mayor deficiencia esquelética (13). En los pacientes cirróticos, se ha visto una mayor reducción en la masa ósea en pacientes con estadíos de enfermedad hepática avanzados, medidos en términos de Child-Pugh (4). En un estudio de la Clínica Mayo, se demostró que la severidad de la enfermedad hepática contribuye significativamente con la pérdida de masa ósea (7), así mismo, en el estudio de Wiboux et al., la DMO de cadera total se correlacionó con el puntaje MELD y la albúmina sérica (8).
Como se ha mencionado previamente, la incidencia de osteoporosis es el doble de alta en los pacientes con enfermedad hepática crónica comparado con la población general (9). La tasa de fracturas vertebrales y no vertebrales se encuentra incrementada en la enfermedad hepática crónica, especialmente en mujeres postmenopáusicas.
Las fracturas periféricas se han relacionado con la cirrosis establecida, el hipogonadismo y el abuso de alcohol. La tasa de fracturas es baja en pacientes no cirróticos eugonádicos (13) La tasa de fracturas incidentes está menos caracterizada, pero la DMO en columna lumbar parece ser un factor de riesgo para incremento en la fractura vertebral (13).
En el estudio de Wiboux et al., que incluyó 99 pacientes, se encontró que, por criterios de la Organización Mundial de la Salud (OMS), el 38% tienen osteoporosis y el 35% tiene osteopenia; el 48% tienen historia previa de fracturas, y el 36% se les documenta al menos una fractura radiológica en cuerpos vertebrales, (algunos presentaron hasta 7 fracturas). La DMO a nivel de columna lumbar, cadera total y cuello femoral se asoció con la presencia de fracturas vertebrales radiográficas. La DMO de cadera total fue un factor de riesgo independiente para la presencia de fracturas radiográficas vertebrales. Los sitios de fracturas por fragilidad clínicas más frecuentemente reportados fueron la columna vertebral (10%), pelvis (4%), radio distal (27%), costillas (21%), fémur proximal (2%), tobillo (10%), tibia (15%), codo (6%), metatarsos (4%) (15).
Los marcadores de recambio óseo se pueden alterar en pacientes con enfermedad hepática avanzada. En el estudio de Wiboux et al., todos los marcadores de recambio óseo se hallaron anormales. Los marcadores de resorción, como los puentes cruzados de colágeno sérico, el fragmento C-terminal de telopéptidos del procolágeno tipo 1 (ICTP) se correlacionaron con la DMO de columna lumbar (15).
No está claramente establecido si el diagnóstico específico de cirrosis es un factor asociado con la salud ósea. Se examinará brevemente el papel que juega cada etiología en el desarrollo de la osteodistrofia hepática.
FISIOPATOLOGÍA DE LA OSTEODISTROFIA HEPÁTICA
La enfermedad hepática, y más precisamente la cirrosis, producen cambios metabólicos complejos que conducen al desarrollo de alteraciones óseas que se conocen como la osteodistrofia hepática. Este fenómeno ha sido estudiado ampliamente, y se ha demostrado que los marcadores de formación ósea están reducidos en pacientes con enfermedad hepática de larga data (4).
En el estudio de Diamond et al., compara la histomorfometría dinámica ósea en pacientes con y sin cirrosis. Encuentran que el volumen y grosor trabecular están significativamente reducidos (p<0,001 hombres y p<0,01 mujeres), que hay hallazgos histológicos compatibles con osteoporosis en el 21% de los sujetos con cirrosis y una disminución de la tasa de formación ósea en el 57%. Concluyen que los pacientes con cirrosis tienen un defecto en la función osteoblástica, dada por una disminución en el grosor del osteoide, de la superficie osteoblástica y la tasa de formación ósea (10).
Los cambios metabólicos complejos de la enfermedad hepática producen alteraciones en el metabolismo óseo; y en general el deterioro de la masa y calidad ósea obedecen a factores múltiples como son las deficiencias nutricionales, el hipogonadismo, el uso de medicamentos, la deficiencia de vitamina D, la inflamación crónica, la hiperbilirrubinemia y la severidad de la enfermedad hepática (Figura 1).

Figura 1: Fisiopatología de la osteodistrofia hepática
Al evaluar la DMO se puede encontrar marcada heterogeneidad en la población de pacientes con cirrosis, que oscila desde ningún efecto hasta el déficit marcado (11). Las variables como índice de masa corporal, historia de ingesta de corticoides, edad y género pueden predecir la presencia de osteoporosis y riesgo de fractura (12).
A continuación, se revisarán algunos de los factores que condicionan el desarrollo de la osteofistrofia hepática (Figura 2).
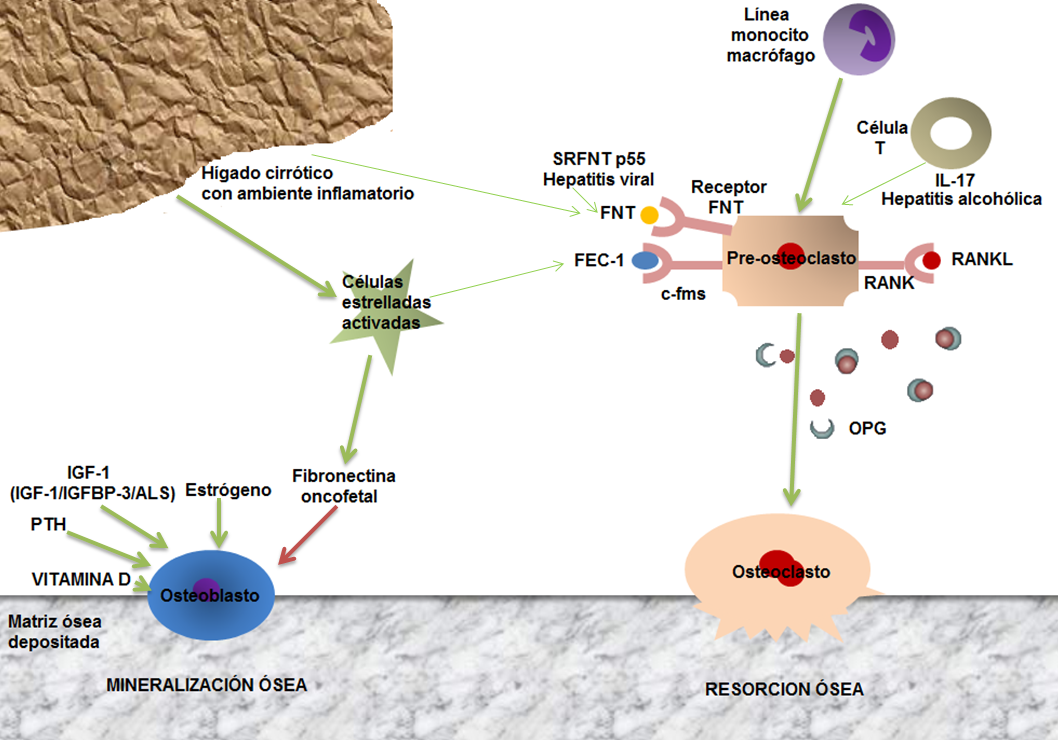
Figura 2: Factores que alteran el recambio óseo en el paciente con enfermedad hepática. El preosteoclasto es activado por las citoquinas inflamatorias como el Factor de Necrosis Tumoral (FNT), el cual está elevado en sujetos con hepatitis viral y alcohólica. El Factor Estimulante de Colonias 1 (FEC-1) se liga a su receptor c-fms para inducir osteoclastogénesis. Los sujetos con enfermedad colestásica tienen FEC-1 elevado promoviendo un ambiente inflamatorio. La Interleuquina 17 (IL-17) es producida por un subgrupo de linfocitos T y está incrementada en hepatitis alcohólica y potencialmente promueve la pérdida ósea. RANKL es un regular clave en la función del osteoclasto y se liga a su receptor RANK para inducir osteoclastogénesis. La osteoprotegerina (OPG) es una receptor soluble que se liga al RANKL produciendo inhibición. La parathormona (PTH), la somatomedina C (IGF-1) y la fibronectina oncofetal afecta la formación del osteoblasto. La fibronectina oncofetal es producida por las células estrelladas y suprime el osteoblasto y la formación ósea en pacientes con cirrrosis biliar primaria. Las flechas verdes son promotoras, las flechas rojas son inhibitorias. (Modificado de: Inaam A et al. Nat. Rev. Gastroenterol. Hepatol. 2009; 6, 660–670)
Desórdenes colestásicos
Los desórdenes colestásicos, como la colangitis esclerosante primaria y la cirrosis biliar primaria, han mostrado una reducción en la DMO en forma acelerada comparada con otras etiologías; sin embargo, la colestasis per se no difiere de otras alteraciones no colestásicas en términos de osteoporosis y riesgo de fractura (13).
En la colangitis esclerosante primaria, el riesgo de fractura está relacionado con el aumento de la edad, la coexistencia de enfermedad inflamatoria intestinal y enfermedad biliar avanzada (14).
La cirrosis biliar primaria ha mostrado una asociación más marcada, seguramente porque afecta mujeres de edad avanzada en postmenopausia (12). El 43% de las mujeres con cirrosis biliar primaria son osteoporóticas y hasta 22% tienen fracturas (7). Las mujeres con cirrosis biliar primaria tienen 4 veces más riesgo de osteoporosis y 2 veces más riesgo de fracturas, cuando son comparadas con controles(14). Se ha demostrado que la disminución de la formación ósea es el principal mecanismo de baja masa ósea en esta forma de enfermedad hepática.
Hepatitis virales
La prevalencia de osteoporosis en hepatitis viral crónica con cirrosis es del 20-53%(15).La cirrosis viral se ha asociado con reducción significativa de la proteína ósea Gla (BGP), hormona paratiroidea (PTH), testosterona en hombres, niveles de 25-OH vitamina D, aumento en la fosfatasa alcalina específica del hueso y el fragmento C-terminal de telopéptido del procolágeno tipo 1(16,17).
Hemocromatosis
Se ha reconocido la presencia de osteoporosis en pacientes con cirrosis asociada a hemocromatosis. Un estudio encontró que el riesgo de osteoporosis es influenciado por la presencia de cirrosis y por el grado de sobrecarga de hierro (18).
Sinigaglia et al. demuestraron que la enfermedad ósea es más común en pacientes con hipogonadismo (22) y que la enfermedad hepática crónica asociada a sobrecarga de hierro usualmente se acompaña de hipogonadismo (19).
Así mismo, se ha encontrado que los niveles elevados de hierro asociados a otras condiciones que presentan sobrecarga de hierro, como son la talasemia y la siderosis, están asociados a osteoporosis (20).
Deficiencia nutricional
Como se evidencia en el trabajo de Cabré y Gassul, las deficiencias nutricionales son comunes en la cirrosis y pueden asociarse la osteodistrofia hepática(21). Así mismo, la pérdida de masa muscular y los bajos índices de masa corporal, asociados a la enfermedad crónica, se han visto involucrados en el desarrollo de anormalidades óseas.
Hipogonadismo
El hipogonadismo se encuentra presente hasta el 75% de los pacientes con cirrosis y se ha visto que juega un papel fundamental en el desarrollo de enfermedad ósea (22),.
En mujeres con enfermedad hepática, se ha encontrado una pérdida de la masa ósea mayor que en controles sanos (13).
Medicamentos
Los medicamentos empleados para el tratamiento de la enfermedad hepática pueden tener efectos adversos en el metabolismo óseo. Entre ellos, los corticoides empleados para el manejo de pacientes con hepatitis autoinmune, la colestiramina, los diuréticos y los antivirales (Figura 3).

Figura 3. Efectos de los medicamentos frecuentemente usados en cirrosis en relación con la masa ósea.
Glucocorticoides
Múltiples condiciones crónicas requieren uso de glucocorticoides a largo plazo como parte fundamental de tratamiento, incluyendo alteraciones hepáticas, gastrointestinales, reumatológicas, renales y pulmonares; como también la supresión para rechazo en trasplante de órganos.
Los corticoides tienen efectos adversos sobre el metabolismo óseo ampliamente estudiados. Por una parte, producen inhibición de la función osteoblástica; por otro lado, reducen la absorción de calcio a nivel intestinal, debido a que interfiere con la acción de la 1,25-OH vitamina D. Así mismo, incrementa la excreción de calcio, induciendo hiperparatiroidismo secundario, y como consecuencia, genera un aumento en la resorción ósea osteoclástica mediante la producción de IL-1. De igual manera, puede asociarse al hipogonadismo secundario (23).
Los glucocorticoides conllevan a catabolismo sistémico y tiene múltiples efectos adversos. La prevalencia de eventos adversos son proporcionales a las dosis. La dosis oral de prednisona mayor o igual a 7,5 mg día produce importantes efectos osteoporóticos (24). Sin embargo, las dosis intermitentes o inferiores a 5 mg día de prednisona, o su equivalente, pueden conferir riesgo incrementado de osteoporosis(25). Los eventos adversos no se reducen por la administración de días alternos (26). Sin embargo, la administración local reduce los eventos adversos sistémicos.
Los glucocorticoides tienen efectos nocivos directos e indirectos sobre el metabolismo óseo. La mayor tasa de pérdida de hueso se da en los primeros seis meses de uso, con promedio de disminución del 5% durante el primer año de tratamiento. Posteriormente se presenta una pérdida de masa ósea entre 1 a 2% anual. El hueso trabecular incluyendo el fémur proximal y el borde cortical de los cuerpos vertebrales son los más susceptibles al efecto deletéreo de los glucocorticoides, comparado con la región cortical de los huesos largos. De este modo la columna lumbar y el fémur proximal son particularmente vulnerables a la pérdida ósea con las fracturas relacionadas por glucocorticoides. Los pacientes jóvenes que reciben glucocorticoides crónicos pierden masa ósea más rápido que los pacientes mayores (27); sin embargo, la mujer postmenopáusica con uso de glucocorticoides a dosis equivalentes tiene mayor riesgo de fractura posiblemente porque se amplifica la perdida ósea. Los efectos esqueléticos de los glucocorticoides son similares para los hombres y así como también para las razas negra y blanca.
El uso de glucocorticoides orales a largo plazo incrementa el riesgo de fractura de cadera y vertebral. La incidencia de fractura asociada a perdida ósea por glucocorticoides se estima entre 1,3 a 2,6 veces más en población que recibe glucocorticoides comparados con aquellos que no reciben. Los pacientes que reciben glucocorticoides a largo plazo, el 30 – 50% desarrollan fracturas. El riesgo de fractura de cadera se duplica y el riesgo de fractura vertebral se incrementa al menos 3 veces comparado a los pacientes que no reciben glucocorticoides.
Los efectos predominantes sobre hueso son la reducción en la formación de hueso por disminución en la replicación y diferenciación del osteoblasto e incremento de la apoptosis.
Los glucocorticoides inhiben la liberación de factores de crecimiento celular como la somatomedina C y el factor de crecimiento transformante beta. La transcripción de los genes de osteoblastos, incluyendo colágeno tipo I y osteocalcina están disminuidos (28). Los glucocorticoides producen atrofia muscular que conlleva a una reducción en la fuerza del estimulo mecánico requerido para estimular la formación de hueso nuevo (29,30).
Los efectos directos e indirectos de glucocorticoides sobre la resorción son menos profundos que sus efectos negativos sobre la formación ósea, pero también contribuye al rápido incremento en el riesgo de fractura (31); promueven la perdida ósea a través la inhibición de la liberación de gonadotrofinas produciendo hipogonadismo; la disminución en número, tiempo de vida y función de los osteoblastos. Adicionalmente, los glucocorticoides promueven la excreción de calcio e inhiben la absorción de calcio promoviendo la hipocalcemia, con el consecuente incremento de la hormona paratiroidea (PTH) e incremento de la resorción ósea(32).
Los pacientes tratados con glucocorticoides tienen factores de riesgo independientes adicionales que conllevan a la pérdida de masa ósea y fracturas; entre ellos, su enfermedad primaria de base, particularmente las alteraciones inflamatorias y autoinmunes que causan detrimento del metabolismo óseo independiente del tratamiento con glucocorticoides.
Colestiramina
Impide la unión de ácidos grados y de esta manera, reduce la absorción gastrointestinal de 25-hidroxivitamina D. Además, la precipitación de sales de calcio a nivel intestinal, por la presencia de grasas no absorbidas, puede contribuir a la mala absorción de calcio.
Diuréticos
El uso de diuréticos es común en pacientes con enfermedad hepática avanzada. Los diuréticos de ASA promueven la pérdida renal de calcio y se asoció con una pérdida pequeña de masa ósea en cadera en una cohorte de 8127 mujeres mayores de 65 años seguidas por más de 4 años; en este estudio no hubo asociación con fractura (33). En una cohorte del WHI (Women’s Health Initiative) con 133855 mujeres seguidas por más de 7 años, se halló que el uso prolongado de diurético se asoció con mayor riesgo de fractura en mujer posmenopáusica, pero no se asoció con alteración de la masa ósea o caídas (34). En un estudio con 3269 hombres mayores de 65 años seguidos por más de 4 años, se halló un incremento en la pérdida de masa ósea(35).
La espironolactona es un diurético antiandrógeno que no tiene estudios de asociación con osteoporosis y sus desenlaces.
Antivirales
Un estudio de corte transversal sugiere que la ribavirina puede inducir pérdida de masa ósea en pacientes con hepatitis crónica (Z-score 1.5 veces disminuido comparado con pacientes no tratados con ribavirina) (36), pero el mecanismo de este efecto no ha sido claramente determinado y ha sido refutado por otro estudio longitudinal (37).
Consumo de alcohol
Los pacientes con cirrosis alcohólica tienen un patrón de bajo recambio óseo. En un estudio de 56 hombres alcohólicos, se demostró que un 32% tenía niveles de 25-OH vitamina D y DMO disminuidas (38). La ingesta excesiva de alcohol puede llevar a un desbalance entre la formación y la resorción ósea, que favorece el desarrollo de osteopenia. Así mismo, el consumo de alcohol es un factor de riesgo independiente para osteoporosis y además incrementa el riesgo de fractura de cadera en 2.8 veces (7).
Deficiencia de vitamina
La literatura reporta que los niveles de vitamina D están disminuidos en 2/3 de los pacientes con cirrosis y en 96% de los pacientes que se encuentran en espera de trasplante hepático (39). En el estudio de Wiboux et al, se reportó un 88% de prevalencia de insuficiencia y deficiencia de vitamina D (15).
El hígado tiene un papel esencial en el metabolismo de la vitamina D. Se encarga de la secreción de sales biliares, la absorción de vitamina D3 de la dieta y la 25-hidroxilación de la vitamina D (40). En pacientes con enfermedad hepática, la colestasis, ciertos medicamentos incluyendo los glucocorticoides, alteran la captación y metabolismo de vitamina D (42).
La osteomalacia es infrecuente en los pacientes con cirrosis (12); sin embargo, la hipovitaminosis D se encuentra asociada con la reducción de la DMO, el bajo recambio óseo y el aumento del riesgo de fracturas (42) que ocurre en estos pacientes.
Inflamación crónica
El proceso inflamatorio crónico, la alteración del flujo sanguíneo hepático, la activación de células estrelladas y la pérdida de la función sintética son algunas de las consecuencias de la cirrosis que se encuentran en relación a las alteraciones metabólicas que ocurren a nivel óseo (Figura 4).

Figura 4. El papel de la inflamación crónica hepática en la activación de la osteoclastogénesis e inhibición del osteoblasto y formación de hueso. FNT (Factor de Necrosis Tumoral); OPG (osteoprotegerina); PTH (parathormona); IGF-1 (Somatomedina C).
El factor de crecimiento similar a la insulina (IGF-1) juega un papel importante en la remodelación ósea y el mantenimiento de la masa ósea. Sin embargo; éste se encuentra disminuido en enfermedad hepática avanzada, de manera proporcional a la severidad de la enfermedad. Se ha demostrado que el reemplazo de IGF-1 en ratas es efectivo para la prevención e incluso la reversión parcial de la osteoporosis (19).
La leptina, producida por las células adiposas, tiene acción periférica para el control de masa ósea mediante el aumento de la proliferación osteoblástica, la síntesis de matriz ósea y la supresión de la producción de RANK-L y por lo tanto disminuye la resorción ósea. Esta molécula se encuentra disminuida en algunas formas de enfermedad hepática, especialmente las colestásicas (19).
Las citoquinas proinflamatorias, como el factor de necrosis tumoral (TNF) y la interleucina 1 (IL-1) estan aumentadas por el proceso inflamatorio hepático y la fibrosis, produciendo un aumento en la actividad de osteoclastos por activación de sus precursores. Estos se encuentran aumentados principalmente en las hepatitis virales y la cirrosis inducida por alcohol (19).
CONCLUSION
En la osteodistrofia hepática el deterioro de la masa ósea es complejo y se debe a factores relacionados con la enfermedad propia, complicaciones nutricionales y medicamentos. Aun no se ha evaluado la carga genética individual en esta patología
RECOMENDACIONES
En el paciente con cirrosis se le debe practicar una DMO.DXA
Realizar una medición sérica de 25.OH vitamina D, niveles de calcio y fósforo séricos
Incentivar el ejercicio y las medidas antifractura
Corregir los factores de riesgo modificables
Corregir los defectos de calcio y vitamina D en los pacientes que reporten alteración
Tratar la osteoporosis y el deterioro de la masa ósea de acuerdo a las recomendaciones actuales para manejo de osteoporosis
BIBLIOGRAFIA
1. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008, 88(1):125-72
2. Friedman SL, Rockey DC, Bissell DM. Hepatic fibrosis 2006: report of the Third AASLD Single Topic Conference. Hepatology 2007, 45(1):242-9
3. Collier, Jane. Bone Disorders in Chronic Liver Disease. Hepatology. 2007;46:1271-1278.
4. Olsson R, Johansson C, Lindstedt G, Mellstrom D. Risk factors for bone loss in chronic active hepatitis and primary biliary cirrhosis. Scand J Gastroenterol 1994;29:753–756.
5. Stellon AJ, Davies A, Compston J, Williams R. Bone loss in autoimmune chronic active hepatitis on maintenance corticoste- roid therapy. Gastroenterology 1985;89:1078 –1083
6. Collier JD, Ninkovic M, Compston JE. Guidelines on the management of osteoporosis associated with chronic liver disease. Gut. 2002;5O(Suppl l):il-i9.
7. Menon,K.v.,Angulo,P.,Weston,S., Dickson, e. R. & Lindor, K. D. Bone disease in primary biliary cirrhosis: independent indicators and rate of progression. J. Hepatol. 35,316–323 (2001).
8. Wibaux C, Legroux-Gerot I, Dharancy S, Boleslawski E, Declerck N, Canva V, Mathurin P, Pruvot FR, Cortet B. Assessing bone status in patients awaiting liver transplantation. Joint Bone Spine 78 (2011) 387–391
9. Diamond T, Stiel D, Lunzer M, et al. Osteoporosis and skeletal fractures in chronic liver disease. Gut. 1990;31:82-87.
10. Diamond TH et al. Hepatic osteodystrophy. Static and dynamic bone histomorphometry and serum bone Gla-protein in 80 patients with chronic liver disease. Gastroenterology. 1989 Jan;96(1):213-21.
11. Guañabens N, Parés A. Liver and bone. Archives of Biochemistry and Biophysics 503 (2010) 84–94
12. American Gastroenterological Association Medical Position Statement: Osteoporosis in Hepatic Disorders. Gastroenterology 2003;125:937–940
13. Janes CH, Dickson ER, Okazaki R, Bonde S, McDonagh AF, Riggs BL. Role of hyperbilirubinemia in the impairment of osteo- blast proliferation associated with cholestatic jaundice. J Clin Invest 1995;95:2581–2586.
14. Guanabens N, Pares A, Ros I, et al.: Severity of cholestasis and advanced histological stage but not menopausal status are the major risk factors for osteoporosis in primary biliary cirrhosis. J Hepatol 2005, 42(4):573–577.
15. Luxon. Bone Disorders in Chronic Liver Diseases. Curr Gastroenterol Rep (2011) 13:40–48
16. Corazza GR, Trevisani F, Di Stefano M, De Notariis S, Veneto G, Cecchetti L, Minguzzi L, Gasbarrini G, Bernardi M. Early increase of bone resorption in patients with liver cirrhosis secondary to viral hepatitis. Dig Dis Sci 2000;45:1392–1399.
17. Tsuneoka K, Tameda Y, Takase K, Nakano T. Osteodystrophy in patients with chronic hepatitis and liver cirrhosis. J Gastroen- terol 1996;31:669–678.
18. Sinigaglia L, Fargion S, Fracanzani AL, Binelli L, Battafarano N,Varenna M, Piperno A, Fiorelli G. Bone and joint involvement in genetic hemochromatosis: role of cirrhosis and iron overload. J Rheumatol 1997;24:1809–1813.
19. Diamond T, Stiel D, Posen S. Osteoporosis in hemochromatosis: iron excess, gonadal deficiency, or other factors? Ann Intern Med 1989;110:430–436.
20. voskaridou, e. & Terpos, e. New insights into the pathophysiology and management of osteoporosis in patients with beta thalassaemia. Br. J. Haematol. 127, 127–139 (2004)
21. Cabre ́ E, Gassull MA. Nutritional and metabolic issues in cirrho- sis and liver transplantation. Curr Opin Clin Nutr Metab Care 2000;3:345–354.
22. Kaymakoglu S, et al. Hypogonadism is not related to the etiology of liver cirrhosis. J Gastro- enterol 1995;30:745-750.
23. Vleggaar FP, van Buuren HR, Wolfhagen FHJ, Schalm SW, Pols HAP. Prevention and treatment of osteoporosis in primary biliary cirrhosis. Eur J Gastroenterol Hepatol 1999;11:617–621.
24. van Staa TP, Leufkens HG, Abenhaim L, et al. Oral corticosteroids and fracture risk: Relationship to daily and cumulative doses. Rhamatology (Oxford). 2000;39: 1383-1389.
25. van Staa TP, Leufkens HG, Abenhaim L, et al. Use of oral corticosteroids and risk of fractures. J Bone Miner Res. 2000;15:993-1000.
26. Ruegsegger P, Medici TC, Anliker M. Corticosteroid induced bone loss. A longitudinal study of alternate day therapy in patients with bronchial asthma using quantitative computed tomography EurJ Clin Pharrnacol. 1983;25:615-620.
27. Lukert BP, Raisz LG. Glucocorticoid-induced osteoporosis: Pathogenesis and management. Ann Intern Med. 1990;112:352-364.
28. Baxter JD. Advances in glucocorticoid therapy Adv Intern Med. 2000;45:317-349.
29. Cunnane G, Lane NE. Steroid-induced osteoporosis in systemic lupus erythematosus. Rham Dis Clin North Am.2000;26:3 1 l-329, vi-vii.
30. Goldstein MF, Fallon JJ Jr, Harning R. Chronic glucocorticoid therapy-induced osteoporosis in patients with obstructive lung disease. Chest. 1999;116:1733-1749.
31. Defranco DJ, Lian JB, Glowacki J 1992. Differential effects of glucocorticoid on recruitment and activity of osteoclasts induced by normal and osteocalcin-deficient bone implanted in rats. Endocrinology 131:114-121
32. Boling, EP. Secondary Osteoporosis: Underlying Disease and the Risk for Glucocorticoid-Induced Osteoporosis. Clin Ther. 2004;26: 1-14)
33. Lim, LS, Fink, HA, Blackwell, T.Taylor,BC. Ensrud, KE. Loop Diuretic Use And Rates Of Hip Bone Loss, And Risk Of Falls And Fractures In Older Women J Am Geriatr Soc. 2009May ; 57(5): 855–862
34. Carbone, LC; Johnson, KC; Bush, AJ; Robbins, J; Larson, JC; Thomas, A; LaCroix, AZ. Loop Diuretic Use and Fracture in Postmenopausal Women. Findings From the Women’s Health Initiative. Arch Intern Med. 2009;169(2):132-140
35. Lim, LS; Fink, HA; Kuskowski, MA; Taylor, BC; Schousboe, JT; Ensrud, KE; for the Osteoporotic Fractures in Men (MrOS) Study Group. Loop Diuretic Use and Increased Rates of Hip Bone Loss in Older Men. The Osteoporotic Fractures in Men Study. Arch Intern Med. 2008; 168 (7):735-740
36. Solis-Herruzo JA, Castellano G, Fernandez I, Munoz R, Hawkins F. Decreased bone mineral density after therapy with alpha interferon in combination with ribavirin for chronic hepatitis C. J Hepatol 2000;33:812–817.
37. Trombetti A, Giostra E, Mentha G, Negro F, Rizzoli R. Lack of evidence for ribavirin-induced bone loss. Hepatology 2002;36: 255–257.
38. Mobarhan SA, Russell RM, Recker RR, Posner DB, Iber FL, Miller P. Metabolic bone disease in alcoholic cirrhosis: a comparison of the effect of vitamin D2, 25-hydroxyvitamin D, or supportive treatment. Hepatology 1984;4:266–273.
39. Crawford BA, Labio ED, Strasser SI, McGaughan GW. Vitamin D re- placement for cirrhosis-related bone disease. Nat Clin Pract Gastroenterol Hepatol 2006;3:689-699
40. Arnaud SB. 25-Hydroxyvitamin D3 treatment of bone disease in primary biliary cirrhosis. Gastroenterology 1982;83:137–140.
41. Wolfhagen FH, van Buuren HR, Vleggaar FP, Schalm SW Management of osteoporosis in primary biliary cirrhosis.Baillieres Best Pratt Res Clin Gastroenterol. 2000;14:629-641.
42. Hay JE. Bone disease in cholestatic liver disease. Gastroenterology 1995;108:276–283
No hay comentarios:
Publicar un comentario